OVERALL SAFETY SUMMARY
In healthy subjects and hospitalized patients with PCR-confirmed SARS-CoV-2 infection, graded elevations in ALT and AST have been observed with a loading dose of remdesivir 200 mg administered intravenously on Day 1 followed by 100 mg administered intravenously once daily for up to 9 days. The mechanism of these elevations is unknown.
Patients should have appropriate clinical and laboratory monitoring to aid in the early detection of any potential adverse events. The decision to continue or discontinue remdesivir after the development of an adverse event should be made based on the clinical risk-benefit assessment for the individual.
Clinical Trials Experience
In a randomized, open-label clinical trial (Study GS-US-540-5773) of remdesivir in 397 subjects with severe COVID-19 treated with remdesivir for 5 (n=200) or 10 days (n=197), adverse events were reported in 71% and 74% of subjects, respectively, serious adverse events were reported in 21% and 35% of subjects, respectively, and Grade ≥3 adverse events were reported in 31% and 43% of subjects, respectively. Nine (5%) subjects in the 5-day group and 20 (10%) subjects in the 10-day group discontinued treatment due to an adverse event. All-cause mortality at Day 28 was 10% vs 13% in the 5- and 10-day treatment groups, respectively.
Hepatic Adverse Reactions
Clinical Trials Experience
Experience in Healthy Volunteers
Grade 1 and 2 transaminase elevations were observed in healthy volunteers in Study GS-US-399-5505 (200 mg followed by 100 mg dosing for 5–10 days) and Study GS-US-399-1954 (150 mg daily for 7 or 14 days), which resolved after discontinuation of remdesivir.
Experience in Patients with COVID-19
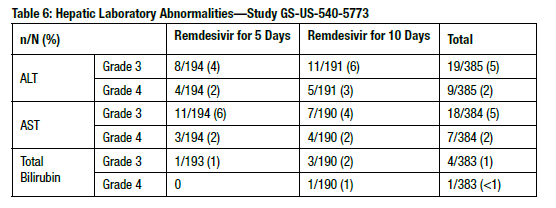
Grade ≥3 hepatic laboratory abnormalities reported in study GS-US-540-5773 of remdesivir in 397 subjects with severe COVID-19 treated with remdesivir for 5 (n=200) or 10 days (n=197) are shown in Table 6.
Experience in Patients with Ebola Virus Disease
In the PALM study, 175 subjects with Ebola virus disease were randomized to receive remdesivir. No SAEs of transaminase elevations or hepatic events were reported. Twenty subjects received remdesivir in a double-blinded, randomized, viral persistence study in the semen of Ebola survivors. Preliminary results indicated there were no SAEs for transaminase elevations.
Compassionate Use Experience
Experience in Patients with COVID-19
In the compassionate use program in patients with severe or critical illness with COVID-19, liver function test abnormalities were reported in 11.7% (19/163) of patients. Time to onset from the first dose ranged from 1-16 days. Four of these patients discontinued remdesivir treatment with elevated transaminases occurring on Day 5 of remdesivir treatment as per protocol.
Seven cases of serious liver-related laboratory abnormalities were identified. There was 1 serious adverse event (SAE) of blood bilirubin increased in a critically ill patient with septic shock and multiorgan failure. None of the other cases had reported adverse events suggestive of hyperbilirubinemia or symptoms of hepatitis.
This website is for Healthcare Professionals only.
Please confirm if you are a healthcare professional.
Disclaimer: Mylan, a Viatris company does not recommend the use of its product Desrem™ (Remdesivir for injection 100mg/vial, hereinafter, the “Product”) in any manner inconsistent with that described in the full prescribing information. The data contained in this microsite is for medical and scientific informational purposes only. Information presented on the microsite should not be construed as advice or recommendation for using the Product. Mylan, a Viatris company expressly disclaims liability for any undue consequences arising out of off-label use of the Product. Mylan, a Viatris company or any of its affiliates, directors, employees, officers, consultants, service providers shall not be responsible in any manner whatsoever, for any action taken, opinions expressed, advice rendered or accepted, any direct, incidental, special or consequential loss and damage caused on the basis of any material or information presented on this microsite. Please refer to full prescribing information before prescribing the Product.